
法洛四联症(tetralogy of Fallot,TOF)是最常见的紫绀型先天性心脏病(congenital heart disease,CHD),占紫绀型CHD的50%~70%[1]。TOF的确切发病机制尚不明确,可能与环境因素和遗传疾病相关。研究表明,染色体非整倍体异常及22q11.2微缺失等拷贝数变异(copy number variations,CNVs)与TOF相关[2, 3]。本研究采用低深度全基因组测序(whole genome sequence,WGS)技术对TOF小儿和胎儿进行分析比较,探讨TOF的遗传学异常,并总结TOF常见合并心内畸形,以期为TOF预后及遗传学咨询提供参考。
资料与方法
一、对象
选取2013年6月至2017年4月于首都医科大学附属北京安贞医院经胎儿超声心动图诊断为TOF并进行WGS的胎儿70例,作为胎儿组,男性39例,女性31例,孕妇初诊孕周19.2~30.4周,平均孕周(24.5±2.7)周。选取2016年2月至2017年9月于首都医科大学附属北京安贞医院行TOF矫治手术并进行WGS的患儿73例,作为小儿组,男性44例,女性29例,年龄0.5~9.6岁,中位年龄0.75(0.66,1.00)岁。本研究为回顾性研究,经首都医科大学附属北京安贞医院伦理委员会批准(批件号:2019030)。所有病例均有遗传学检测知情同意书。
二、方法
1. 超声心动图检查:胎儿超声心动图检查应用GE Voluson E8、E10型彩色多普勒超声诊断仪,2D或3D容积探头,频率4~8 MHz。按照美国超声心动图协会推荐的规范化指南[4]及《胎儿心脏病产前超声诊断咨询及围产期管理指南》[5]对胎儿进行检查。胎儿超声心动图诊断的TOF及胎儿畸形均经出生后复查或终止妊娠后病理解剖确诊。小儿超声心动图检查应用Philip iE33超声诊断仪,使用S5-1探头,频率1~5 MHz。探查小儿左心室长轴切面、大动脉短轴切面、心室短轴切面、四腔心切面、五腔心切面、三腔心切面、两腔心切面、剑下切面、胸骨上窝等切面,测量各切面的心脏参数,二维图像结合彩色多普勒及频谱多普勒探查心脏结构有无异常。小儿TOF病例均行手术并诊断明确。
2. 遗传学样本获取:胎儿经超声心动图诊断心脏畸形,部分孕妇经多学科咨询预后情况后终止妊娠并签署胎儿尸检及遗传学检测知情同意书。终止妊娠病例取脐带组织,出生病例取外周血或指血,同时取父母双方外周血各5 ml(乙二胺四乙酸抗凝),立即置于-80 ℃冷冻,并送基因检测公司进行遗传学检测。小儿TOF经超声心动图诊断明确后,签署遗传学检测知情同意书,提取收集患儿外周血5 ml,送检流程同胎儿。
3. 遗传学检测:使用QIAamp DNA试剂盒提取样本中的基因组DNA(gDNA),采用全基因组低覆盖度测序(Illumina Hiseq2500,测序类型:PE50 bp)方法,检测>100 kb的CNVs,并对已知致病和疑似致病CNVs的家系进行验证。参照美国医学遗传学会解读流程[6],将以上变异的临床意义划分为已知致病、疑似致病、临床意义未明、疑似良性及良性5类。本组病例将已知致病及疑似致病变异确定为致病性变异,即遗传结果阳性。
三、统计学分析
应用SPSS 22.2软件进行统计学分析,年龄为不符合正态分布的计量资料,以M(P25,P75)表示,孕周等数据为符合正态分布的计量资料,以
±s表示。计数资料采用例(%)表示,2组间染色体异常检出率比较采用χ2检验或Fisher确切概率法,以P<0.05为差异有统计学意义。
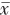
结果
一、胎儿组与小儿组合并的其他心内畸形
胎儿组TOF70例及小儿组TOF73例中,合并其他心内畸形者所占比例分别为70.0%(49/70)、67.1%(49/73)。胎儿组主要合并畸形为动脉导管异常(24/70,34.3%)、右位主动脉弓(16/70,22.8%)、永存左上腔静脉(8/70,11.4%)。小儿组主要合并畸形为右位主动脉弓(12/73,16.4%)、房间隔缺损(12/73,16.4%)、永存左上腔静脉(10/73,13.7%)。143例TOF中,合并的心内畸形最常见为右位主动脉弓(28/143,19.6%)。胎儿组与小儿组的区别主要在于胎儿组易合并动脉导管异常(动脉导管缺如13例、动脉导管细窄或闭锁9例、动脉导管异位2例),而小儿组仅合并动脉导管未闭9例。此外,小儿组中还合并卵圆孔未闭16例(表1 ,图1 、2 )。
表1 143例TOF合并心内畸形情况(例) |
| 心内合并畸形 | 胎儿组(n=70) | 小儿组(n=73) |
|---|---|---|
| 主动脉弓及头臂分支异常 | ||
| 右位主动脉弓+头臂镜像分支 | 11 | 12 |
| 右位主动脉弓+迷走左锁骨下动脉 | 5 | 0 |
| 迷走右锁骨下动脉 | 1 | 1 |
| 双主动脉弓 | 0 | 1 |
| 动脉导管异常 | ||
| 动脉导管缺如 | 13 | 0 |
| 动脉导管细窄或闭锁 | 9 | 0 |
| 动脉导管异位 | 2 | 0 |
| 动脉导管未闭 | 0 | 9 |
| 静脉异常 | ||
| 永存左上腔静脉 | 8 | 10 |
| 无名静脉弓下走行 | 1 | 2 |
| 其他动脉异常 | ||
| 细小冠状动脉-肺动脉瘘 | 0 | 1 |
| 单冠状动脉 | 0 | 1 |
| 右肺动脉异常起源于升主动脉 | 0 | 1 |
| 左侧椎动脉起自于主动脉弓 | 0 | 1 |
| 瓣膜异常 | ||
| 瓣膜黏液样变 | 1 | 0 |
| 二尖瓣闭锁 | 1 | 0 |
| 主动脉瓣二叶畸形 | 1 | 0 |
| 其他 | ||
| 卵圆孔未闭 | 0 | 16 |
| 房间隔缺损 | 0 | 12 |
| 房室间隔缺损 | 2 | 0 |
| 肌部室间隔缺损 | 0 | 5 |
| 心房反位 | 0 | 1 |
注:TOF为法洛四联症 |
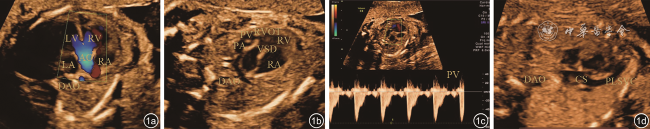
图1 法洛四联症胎儿超声心动图图像。图a为心尖五腔心切面示主动脉增宽,骑跨于室间隔之上,骑跨率约50%,彩色多普勒示双心室血液入主动脉;图b为大动脉短轴切面示室间隔缺损膜周部,室上嵴前移,致右心室流出道狭窄,肺动脉狭窄;图c示肺动脉瓣上流速;图d示永存左上腔静脉汇入冠状静脉窦注:LV为左心室;RV为右心室;AO为主动脉;DAO为降主动脉;VSD为室间隔缺损;PV为肺动脉瓣;PA为肺动脉;RA为右心房;LA为左心房;RVOT为右心室流出道;CS为冠状静脉窦;PLSVC为永存左上腔静脉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
二、胎儿组与小儿组合并的遗传学异常
胎儿组中,遗传检测结果异常21例(21/70,30.0%)。其中,非整倍体5例(5/70,7.1%),包括18-三体2例(2/70,2.9%)、21-三体2例(2/70,2.9%)及13-三体1例(1/70,1.4%)。致病性CNVs 16例(16/70,22.9%),包括22q11.2微缺失综合征12例(12/70,17.1%)、17p11.2微缺失2例(SmithMagenis综合征)(2/70,2.9%)、1p36微缺失1例(1/70,1.4%)、22q11.2微重复1例(1/70,1.4%)。
小儿组中,未检测到非整倍体异常。检测到致病性CNVs 8例(8/73,11.0%),包括22q11.2微缺失综合征5例(5/73,6.8%)、22q11.2 微重复2例(2/73,2.7%)、Xp22.2微重复1例(1/73,1.4%)。
胎儿组与小儿组的染色体异常整体检出率差异存在统计学意义(30.0% vs 11.0%,χ2=8.014,P=0.005),但致病性CNVs检出率差异无统计学意义(22.9% vs 11.0%,P=0.057)。
三、遗传学异常者合并的其他心内畸形
胎儿组中21例遗传学异常者合并的其他心内畸形主要为右位主动脉弓(7/21,33.3%)、动脉导管异常(5/21,23.8%)、永存左上腔静脉(4/21,19.0%)。小儿组中8例致病性CNVs合并的其他心内畸形主要为卵圆孔未闭/房间隔缺损(4/8,50.0%)、右位主动脉弓(3/8,37.5%)、肌部室间隔缺损(3/8,37.5%)。
四、TOF胎儿结局
本研究中70例胎儿TOF,终止妊娠56例,其中21例合并染色体异常;10例肺动脉闭锁合并体肺侧支血管供血;7例肺动脉瓣缺如综合征;18例肺动脉发育尚可,但家属要求终止妊娠。出生14例中,11例完成TOF矫治手术,3例失访。11例手术患儿中,8例因肺动脉发育较好完成一期根治手术,效果良好;3例因肺动脉发育较差,进行了二次手术。
讨论
一、TOF合并心内畸形的特征
TOF属于复杂性CHD,同时容易合并其他心内畸形。既往研究显示[8],57%的TOF患者可合并其他心脏畸形,较常见的畸形包括右位主动脉弓、房间隔缺损或卵圆孔未闭、永存左上腔静脉、房室间隔缺损、冠状动脉解剖异常、多发室间隔缺损等。本研究结果与既往研究基本符合,证实胎儿及小儿TOF最常见的合并畸形为右位主动脉弓,检出率为19.6%(28/143)。
本研究中胎儿组动脉导管异常较常见,表现为动脉导管缺如、狭窄或闭锁及异位,而小儿组表现为动脉导管未闭。房间隔缺损及卵圆孔未闭在小儿组也较常见,原因可能在于产前易受卵圆孔开放影响,对房间隔异常检出困难。因此,除右位主动脉弓外,TOF也容易合并动脉导管及房水平异常的问题,产前及产后超声应多角度探查避免漏诊。
二、TOF合并染色体异常的特征
本研究在胎儿组中发现非整倍体包括21-三体、18-三体及13-三体,而小儿组未检测到非整倍体,主要原因在于非整倍体在产前常规筛查中基本可以检出,且家属一般选择终止妊娠。通常,当合并非整倍体时,患儿的存活率很低。
本研究在小儿组中检测到1例疑似致病CNVs,为Xp22.2重复。这个区域重复与CHD的关系尚未见报道。根据检测到的重复区域涉及范围,该CNVs有可能是先证者的致病原因,但其与CHD的关系仍需进一步研究。
三、心内畸形及遗传学异常对TOF预后的影响
通常,TOF胎儿宫内可以存活,出生后总体预后良好。TOF的预后不仅取决于病变的严重程度,所合并心内其他畸形以及是否存在染色体异常也会影响到手术效果和长期预后。
本研究中最常见的合并心内畸形是右位主动脉弓。胎儿时期,右位主动脉弓与左位导管包绕气管形成“U”型血管环,这种血管环较疏松,虽然包绕气管,但为开放性,导致气管狭窄的可能性较低,大部分不需要手术治疗。但不除外宫内压迫气管可能,若气管受压,则可能会导致气管发育异常以致出生后出现呼吸困难症状。本研究小儿组合并右位主动脉弓病例12例均未出现气管发育异常。
本研究尚存在一定局限性:由于本中心为胎儿心脏病转会诊中心及小儿心脏病诊疗中心,入组病例属于选择性人群,且研究结果为单中心数据,因此可能存在一定的偏倚。此外,本研究只分析了非整倍体及致病性CNVs,大量临床意义不明的CNVs仍需要进一步研究。
综上所述,TOF易合并其他心内畸形及遗传学异常。合并心内畸形以右位主动脉弓异常较多见,产前及产后超声应仔细探查,避免漏诊;合并染色体异常以非整倍体和致病性CNVs为主,一旦确诊建议完善遗传学检测。超声心动图结合WGS可为临床TOF产前产后一体化管理以及预后咨询提供可靠依据。