
女性下生殖道畸形的诊治是国际妇产科学界研究的热点及难点,尤其是青少年女性[1]。MRKH综合征(Mayer-Rokitansky-Küster-Hauser syndrome)是青少年女性常见的下生殖道畸形。其是由于双侧副中肾管(苗勒管)发育、融合障碍导致子宫和阴道中上段发育不全;而染色体、性腺、第二性征及阴道前庭均为正常女性特征的先天性发育异常,常合并泌尿系统和骨骼系统畸形[2]。既往对MRKH综合征影像学的研究主要为MRI。超声虽然是女性生殖道影像学检查的首选方法[3],但由于超声医师对MRKH综合征的认识不够,常无法正确判断MRKH综合征两侧始基子宫,甚至无法准确定位卵巢,更无法确定有无阴道结构,因此易导致临床误诊误治。本研究通过回顾性分析经手术证实的一组MRKH综合征的术前超声表现,并与手术结果进行对比,总结MRKH综合征典型和非典型的超声特征,以期为临床诊治MRKH综合征提供可靠的影像学依据。
材料与方法
一、对象
回顾性分析2021年1月至12月在深圳市罗湖区人民医院超声科检查并经腹腔镜手术证实的75例MRKH综合征患者的超声表现和临床资料,患者年龄15~35(24.16±4.83)岁,排除既往于其他医院行手术治疗和阴道顶压的患者。入组患者临床症状均表现为原发性闭经及性生活困难(有性生活史者),乳腺和外阴发育均正常,其中6例有周期性腹痛,性激素六项检查均正常,其中66例染色体核型检查为46,XX。本研究经深圳市罗湖区人民医院伦理委员会批准(批件号:2021-LHQRMYY-XJSLL-003)。
二、仪器与方法
(一)仪器
应用Mindray Nuewa R9彩色多普勒超声诊断仪,配备SC5-1U腹部凸阵探头(频率1~5 MHz)、L14-5WU浅表线阵探头(频率5~14 MHz)、DE10-3WU腔内容积探头(频率3~10 MHz)和ELC13-4U腔内双平面探头(频率4~13 MHz)。
(二)方法
所有检查均由接受培训且具有5年以上妇产科超声检查经验的医师完成。所有患者均行经腹、经直肠及经直肠双平面超声检查,图像均留存工作站以备分析研究。
1. 经腹及经直肠超声检查:观察盆腔内的双侧始基子宫,并确定有无宫颈和宫腔内膜;当盆腔内未发现始基子宫时,可用高频线阵探头在腹股沟区扫查寻找始基子宫。测量双侧始基子宫的长径和厚度;观察连接双侧始基子宫下缘条带状低回声的索状带,并测量中央连接处的厚度;在两侧始基子宫旁观察同侧卵巢,当盆腔内未发现卵巢时,可在上腹部扫查或使用高频线阵探头在腹股沟区扫查寻找卵巢。观察泌尿系统。
2. 经直肠双平面高频超声检查[4]:启用高频线阵模式观察膀胱尿道后壁与直肠前壁之间的间隙有无阴道结构,以清晰显示尿道全长、膀胱颈、膀胱后壁及会阴体等结构为图像采集标准,并于尿道内口水平测量膀胱尿道后壁与直肠前壁之间间隙的厚度,测量3次,记录平均值。切换至高频凸阵模式,探头在缓慢退出直肠的过程中,连续横切面观察膀胱尿道后壁与直肠前壁之间有无阴道结构。
三、统计学分析
采用SPSS 25.0统计软件分析数据。计量资料以
±s表示,计数资料以例(%)表示。
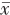
结果
一、超声诊断MRKH综合征与手术的符合率
本组75例MRKH综合征患者均经腹腔镜手术证实,均行经腹腔镜下腹膜代阴道成形术,其术前超声检查结果均与手术结果一致,诊断符合率为100%。
二、MRKH综合征典型的“四联征”超声表现
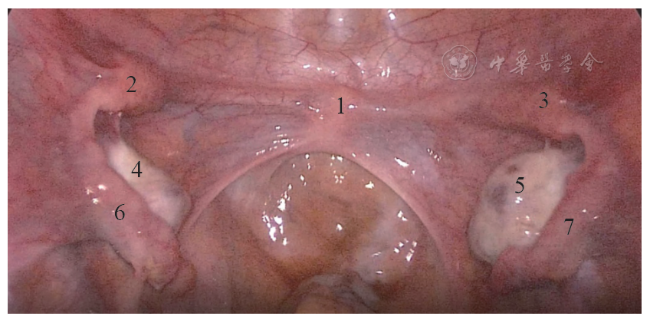
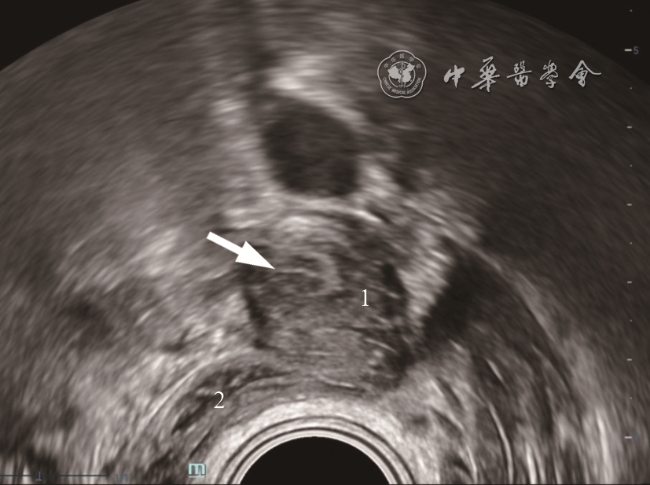
75例MRKH综合征患者中,62例(82.7%)表现为以下典型的“四联征”超声特征(图1 ):(1)双侧始基子宫位于盆腔左右两侧,表现为梭形或长条形肌性回声,未见宫颈和宫腔内膜回声(图2a ,2b ),左侧始基子宫平均长径约(28.44±7.31)mm,平均厚度约(16.22±4.55)mm;右侧始基子宫平均长径约(26.6±7.15)mm,平均厚度约(17.13±5.25)mm;(2)连接两侧始基子宫下缘的索状带位于膀胱底部后下方,表现为条带状低回声(图2c ),中央连接处平均厚度为(7.65±2.69)mm;(3)双侧卵巢分别紧邻同侧始基子宫(图2d);(4)在膀胱尿道后壁与直肠前壁之间未见正常阴道结构,仅见低回声结缔组织(图2e),此间隙在尿道内口水平平均厚度为(3.24±1.87)mm。上述典型超声表现与手术结果一致(图3 )。
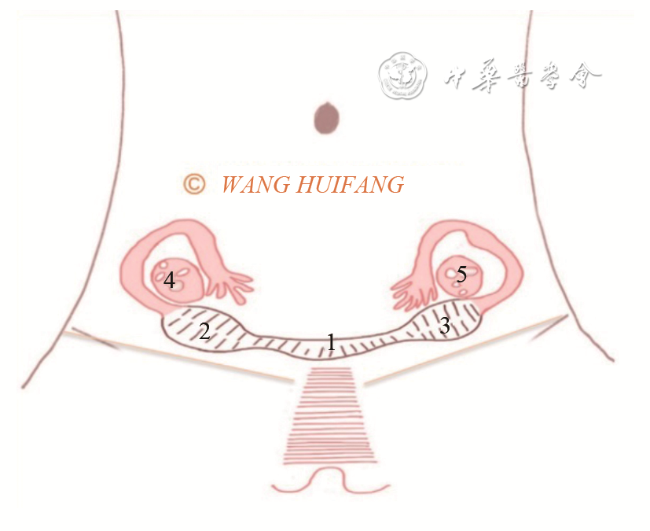
图2 典型MRKH综合征超声图像。图a为经直肠超声显示右侧始基子宫和索状带;图b 为经直肠超声显示左侧始基子宫和索状带;图c 为经直肠超声显示索状带(箭头所示);图d 为经直肠超声显示紧邻左侧始基子宫的卵巢;图e 为经直肠双平面高频超声未见正常阴道结构,仅见低回声结缔组织(箭头所示为尿道内口;虚线所示为结缔组织)注:1 为右侧始基子宫;2 为左侧始基子宫;3 为索状带;4 为卵巢;BL 为膀胱;U为尿道;PB为会阴体 |
三、MRKH综合征非典型的超声表现
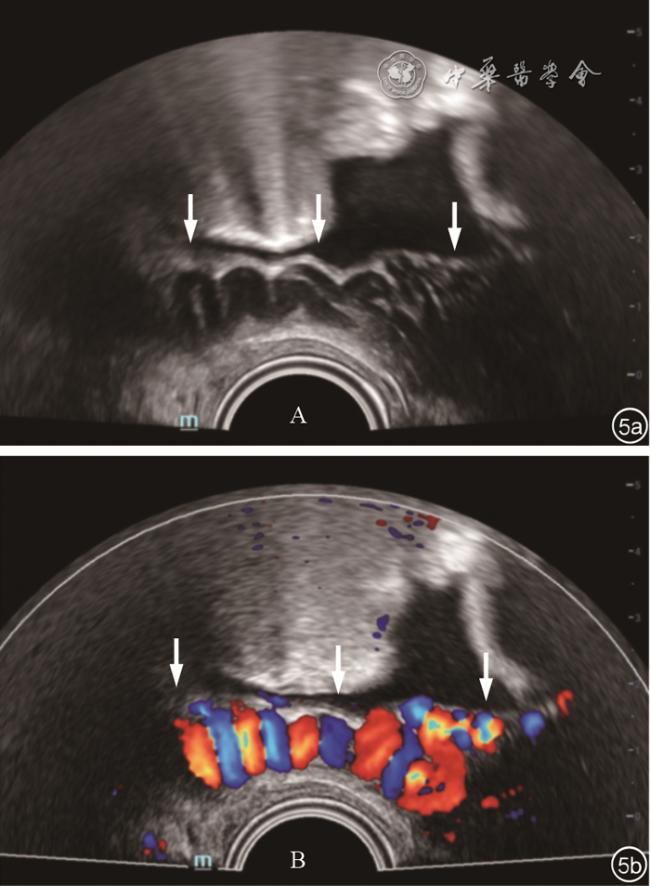
75例MRKH综合征患者中,13例(17.3%)表现为以下非典型的超声特征:(1)始基子宫位置异常,位于腹股沟区,3例单侧和1例双侧;(2)始基子宫有功能性子宫内膜回声(图4 ),3例为单侧,2例为双侧;(3)2例索状带二维超声表现为网状结构(图5a ),彩色多普勒血流表现为扩张的血管(图5b ),频谱多普勒表现为静脉血流频谱;(4)卵巢位置异常,2例双侧卵巢位于上腹部,1例右侧卵巢位于上腹部,1例左侧卵巢位于腹股沟区。其中1例单侧始基子宫合并同侧卵巢疝入腹股沟管,1例索状带静脉扩张合并双侧卵巢位于上腹部。上述超声表现均与手术结果一致。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
四、其他伴发的异常超声表现
75例患者中,一侧始基子宫肌瘤2例,双侧始基子宫腺肌病3例;卵巢畸胎瘤3例,多囊卵巢综合征5例,卵巢囊肿10例。
五、泌尿系统超声表现
75例患者中,单侧肾缺如3例,单侧盆腔异位肾1例。
讨论
MRKH综合征为双侧副中肾管未发育或其尾端发育停滞而未向下延伸所致的以始基子宫、无阴道为主要临床表现的综合征,其病因及发病机制尚不明确[5]。病理学主要表现为苗勒管遗迹(盆腔两侧始基子宫及连接两者下缘的索状带)、双侧卵巢紧邻同侧始基子宫以及上2/3阴道缺如[2]。基于是否合并泌尿系统或骨骼系统发育畸形,国内外专家共识[3,5]将MRKH综合征分为Ⅰ型和Ⅱ型,Ⅰ型即单纯型,单纯子宫、阴道发育异常,而泌尿系统、骨骼系统发育正常,此型常见;Ⅱ型即复杂型,除子宫、阴道发育异常外,还伴有泌尿系统或骨骼系统发育畸形。MRKH综合征的MRI影像学特征可分为典型与非典型表现,以此为依据有利于病情评估及指导后续临床治疗[6]。但目前尚未见对MRKH综合征的超声表现进行分型的研究报道。MRKH综合征为罕见病,发病率为1/5000~1/4000[7],但由于我国人口基数大,因此此类患者在临床工作中并不少见[1]。深圳市罗湖区人民医院女性生殖道畸形与青少年妇科为全国女性生殖道畸形诊治中心之一,为本研究提供了充足的病例来源。本研究对75例经手术证实的MRKH患者的术前超声表现与手术结果进行对比,结果显示诊断符合率为100%。同时,对纳入病例的超声特征进行分析,认为可将其分为典型与非典型的超声表现,其中62例(82.7%)具有典型的“四联征”超声表现:双侧梭形或长条形肌性回声的始基子宫,连接两始基子宫下缘条带状低回声索状带,双侧卵巢紧邻同侧始基子宫及膀胱尿道后壁与直肠前壁之间未见正常阴道结构、仅见低回声结缔组织,上述特征符合MRKH综合征典型的子宫阴道发育异常的病理学特征[2]。
女性生殖道发育过程中,由于各种内源性及外源性因素的影响[1],副中肾管停止发育的程度和时间不一致,可出现非典型的影像学表现[6]。文献报道约10%的MRKH综合征患者始基子宫内有功能性子宫内膜[8],本研究有3例单侧和2例双侧始基子宫有功能性子宫内膜,其中1例年仅15岁,因其左侧始基子宫发育较好且有子宫内膜及宫腔积血,较早出现周期性腹痛,出现梗阻症状,故需尽早手术切除。索状带为苗勒管遗迹中未发育宫颈的原基,其主要解剖结构为疏松结缔组织和静脉血管[2]。盆腔静脉血管壁薄弱、弹性差,部分女性存在卵巢静脉瓣膜缺失和功能不全等解剖变异,因此导致盆腔静脉压增高,继而发生盆腔静脉迂曲扩张[9],索状带内静脉也随之扩张,失去典型的条带状低回声特征。本研究2例患者在连接两侧始基子宫的索状带走行区域二维超声表现为网状结构,彩色多普勒血流显像及频谱多普勒显示为扩张的静脉,此时不可误认为是单纯盆腔静脉曲张,需要沿着曲张的静脉追踪扫查至始基子宫以确认为索状带。超声准确识别索状带静脉扩张,可为预防术中出血提供参考。
卵巢起源于生殖嵴,最初位于后腹壁的上方,在引带的作用下迁移至骨盆,生殖嵴生长受阻或下降障碍可导致卵巢位置异常。卵巢的迁移过程依赖于苗勒管的正常发育[10],苗勒管发育、融合障碍,可导致卵巢位置过高,停滞在腹腔,从而出现卵巢异位的现象,本组病例中3例卵巢位于上腹部。子宫阔韧带或卵巢悬韧带原发性薄弱延长,导致卵巢过度活动,加上始基子宫未融合,则同侧始基子宫及卵巢疝入腹股沟管的可能性会增加[11]。本研究中有1例卵巢与同侧始基子宫疝入腹股沟管,位于腹股沟管的卵巢有扭转和梗死的风险[12]。当在始基子宫旁未发现卵巢时,应在包括腹部和腹股沟区的范围内仔细扫查寻找卵巢。超声准确评估卵巢及其位置对于MRKH综合征患者的不孕症治疗、合并卵巢肿瘤及扭转、盆腔疼痛评估和术前手术方案的制定均具有重要临床意义[10]。
超声评估MRKH综合征应与阴道闭锁Ⅱ型和完全性雄激素不敏感综合征(complete androgen insensitivity syndrome,CAIS)相鉴别。阴道闭锁Ⅱ型(即阴道完全闭锁)为泌尿生殖窦及苗勒管末端发育异常而未形成贯通的阴道所致,虽然亦表现为原发闭经,但子宫体发育正常或虽有畸形但子宫内膜有功能,多合并子宫颈发育异常,往往以生殖道梗阻为主要表现[5,8]。CAIS也表现为原发闭经,但染色体核型为46,XY,性腺为睾丸,但生殖道、泌尿生殖窦和第二性征的发育却具有女性特征或兼具两性的特征;睾丸位于腹腔、腹股沟管或阴唇内,常被误诊为发育不良的始基子宫或前列腺;可见阴道下段[15]。
MRI被认为对于MRKH综合征具有精确的诊断价值[3,5],主要在于MRI能够评估始基子宫有无功能性子宫内膜、索状带及膀胱尿道后壁与直肠前壁之间间隙的形态。近年来,随着超声技术发展,其优势逐渐显现。超声不仅能很好地分辨始基子宫有无功能性子宫内膜,还能大范围扫查异位的始基子宫和卵巢;彩色多普勒超声可显示索状带内扩张的静脉血管;而且本研究团队证实了经直肠双平面高频超声能准确有效地评估阴道形态或膀胱尿道后壁与直肠前壁之间间隙的形态[4]。因此,笔者认为超声与MRI一样可用于MRKH综合征术前的全面精确的评估,但需掌握MRKH综合征典型和非典型的超声表现,以减少漏诊误诊的情况;且超声具有MRI无法比拟的简便、易行、低价和可重复的优势。
综上所述,大部分MRKH综合征具有特征性的“四联征”超声表现,术前超声可明确诊断。对于MRKH综合征非典型的超声表现,联合多种探头多种途径综合评估也可达到精确诊断的目的。超声可为MRKH综合征患者临床手术方案的制定提供可靠的影像学诊断依据。