
22q11.2 微缺失综合征(22q11.2 microdeletion syndrome,22q11.2DS)是人类最常见的微缺失综合征[1]。据报道22q11.2DS 的胎儿发生率为1/992,其活产儿发生率为1/2148,其流产儿发病率约1/1500[2-4]。22q11.2DS 约90% 为新发突变, 约10%遗传自父母,属常染色体显性遗传病,母源性较为常见[5],且与父母年龄无关。
22q11.2DS 的产前诊断对孕期管理及准父母心理准备有重要意义[6]。当22q11.2DS 胎儿发现复杂的先天性心脏病(congenital heart disease,CHD)或其他相关的先天性异常时,可以制定分娩计划,并为母、婴提供适当的生命支持,适时转诊到更高级别的具备新生儿心脏手术及重症监护的医院。当22q11.2DS 胎儿出现免疫功能障碍和低钙血症时,可以提前预见并能得到及时解决[7-8],避免产后还不能及时明确诊断,有效降低发病率和死亡率。产前超声筛查包括早孕期筛查、软指标筛查及中预期胎儿结构筛查,是胎儿结构畸形及染色体软指标的有效检测手段,尤其对于22q11.2DS 常合并的CHD 和颜面部、胸腺、泌尿系统及骨骼畸形等。本文回顾分析12 例产前确诊的22q11.2DS胎儿超声影像学特征,以提高产前22q11.2DS 诊断率。
资料与方法
一、对象
回顾分析2019 年6 月1 日至2022 年5 月31日南京医科大学附属苏州医院优生优育中心实验室通过染色体微阵列(芯片)分析(chromosomal microarray analysis,CMA)确诊的12 例22q11.2DS胎儿。以上所有孕妇均完成产前三级筛查及四级胎儿超声心动图检查,所有阳性病例均经2 名高年资医师签名出具超声报告,并存图入档。
二、仪器与方法
1. 超声仪器:美国通用公司Voluson E10 超声彩色多普勒诊断仪,2D/3D 容积探头,频率4~8 MHz;韩国三星公司WS80A 超声彩色多普勒诊断仪,2D/3D 容积探头,频率4~8 MHz。
3. 介入性产前诊断:介入性产前诊断前充分告知孕妇及家属介入性产前诊断的感染、流产风险及CMA 检测的优点及缺点,征得患者同意后签署手术知情同意书。所有孕妇均在超声引导下行羊膜腔穿刺,抽取10 ml 羊水进行CMA 检测,CMA检测采用美国Affy metrix 公司的单核苷酸多态性微阵列检测平台的高分辨全基因组CytoScan 750K芯片,取250 ng 标本DNA,按照标准操作流程经消化、连接、聚合酶链式反应(polymerase chain reaction,PCR)、PCR 产物检测、纯化、定量、片段化、片段化质控电泳检测、标记、杂交、洗脱、染色、扫描后进行结果分析。数据分析使用配套的软件。
三、统计学分析
运用IBM SPSS Statistics 25 统计软件进行统计学分析,计数资料用百分比、率及频数表示。
结 果
一、一般特征
本组12 例22q11.2DS 胎儿均为单胎。孕妇年龄范围23~41 岁,平均年龄为29 岁,孕周范围18+5~26+1 周,平均孕周为22+1 周;平均怀孕次数2 次,范围1~4 次;平均分娩次数1 次,范围1~3 次。胎儿性别:男性8 例(66.7%),女性4 例(33.3%)。产前介入取样方式:绒毛膜穿刺0 例,羊水穿刺9 例(75.0%),脐带血穿刺3例(25.0%)。家族遗传2 例(16.7%),基因突变10 例(83.3%)。3 例(25.0%)胎儿因产前无结构异常而正常分娩,其余9 例(75.0%)胎儿终止妊娠。
二、超声特征
心脏畸形是22q11.2DS 最常见的异常,本组病例中单纯心脏畸形5 例(41.7%),合并心外畸形3例(25.0%),总计8 例(66.7%)。从超声征象看,室间隔缺损(ventricular septal defect,VSD)5 例(41.7%),胎儿四腔心切面及左心室流出道切面显示室间隔连续性中断,彩色多普勒血流成像可见穿隔的彩色血流信号;胎儿主动脉骑跨于室间隔之上2 例(16.7%);肺动脉狭窄或闭锁4 例(33.3%),可见肺动脉瓣回声增强增厚,启闭活动受限或闭锁,彩色多普勒血流成像可见肺动脉的流速增快,肺动脉闭锁胎儿可见动脉导管的逆灌血流信号;胎儿心轴左偏3 例(25.0%),未探及胎儿心轴右偏。但从心脏复合畸形角度看,圆锥动脉干畸形占7例(58.3%),其中法洛四联症(tetralogy of fallot,TOF)2 例(图1),室间隔缺损型肺动脉闭锁2例,室间隔缺损合并肺动脉狭窄2 例,右心室双出口1 例。右位主动脉弓合并左锁骨下动脉迷走2 例(16.7%),主动脉弓离断1 例(8.3%)。
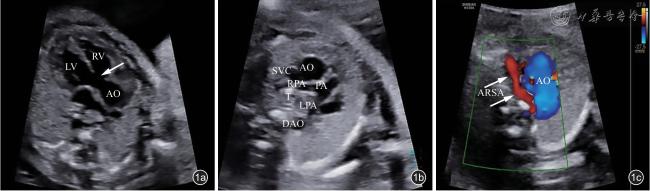
在心外畸形中最常见的是胸腺发育不良3 例(25.0%),其中2 例合并心脏畸形,1 例为单纯性胸腺发育不良。泌尿系统畸形中1 例为多囊肾同时合并长骨发育偏短。
此外在本组病例中3 例(25.0%)胎儿产前未发现明显畸形。
三、基因遗传学检测结果
表1 22q11.2 微缺失综合征病例资料统计表 |
| 序号 | 孕周(周) | 临床指征 | 胎儿性别 | 遗传学检测结果 | 异常区带 | 超声表现 | 结局 |
|---|---|---|---|---|---|---|---|
| 1 | 24+6 | 超声异常 | 女 | 胎儿染色体22q11.21区域存在3.169Mb的DNA片段缺失。ClinGen数据库提示该片段包含22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672),具有明确单倍剂量致病效应(Haploinsuきciencyscore:3) | arr[hg19]22q11.21(18,631,364-21,800,471)x1 | 心轴左偏、膜周部室间隔缺损,肺动脉轻度狭窄,胸腺发育不良 | 终止妊娠 |
| 2 | 20 | 父母一方染色体异常携带 | 男 | 胎儿染色体22q11.21区域存在3.152Mb的片段缺失,涉及46个OMIM基因。Clingen数据库提示该缺失包含22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672),具有明确单倍剂量致病效应(Haploinsufficiencyscore:3),且>90%的缺失为新发变异 | arr[hg19]22q11.21(18,648,855-21,800,471)x1 | 室间隔缺损、主动脉骑跨、肺动脉狭窄、永存左上腔 | 终止妊娠 |
| 3 | 21+2 | 产前筛查高风险超声异常 | 男 | 胎儿染色体22q11.21区域存在3.152Mb的片段缺失。ClinGen数据库提示该片段包含22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672)(Haploinsufficiencyscore:3),且>90%的缺失为新发变异 | arr[hg19]22q11.21(18,648,855-21,800,471)x1 | 右心室双出口、室间隔缺损(主动脉瓣下型)、肺动脉狭窄瓣 | 终止妊娠 |
| 4 | 19 | 超声异常 | 男 | 胎儿染色体22q11.21区域存在3.152Mb的片段缺失。ClinGen数据库提示该片段覆盖22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672)(Haploinsufficiencyscore:3),且>90%的缺失为新发变异。 | arr[hg19]22q11.21(18,648,855-21,800,471)x1 | 胸腺发育不良 | 终止妊娠 |
| 5 | 21 | 产前筛查高风险 | 男 | 胎儿染色体22q11.21区域存在3.152Mb的DNA片段重复,位于22q11微重复综合征致病区域,包含转录因子TBX1等重要基因。ClinGen数据库提示22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672)具有明确三倍剂量致病效应(Triplosensitivityscore:3) | arr[hg19]22q11.21(18,648,855-21,800,471)x3 | 正常 | 正常分娩 |
| 6 | 19+6 | NIPT异常 | 男 | 胎儿染色体22q11.21区域存在3.152Mb的DNA片段重复,位于22q11微重复综合征致病区域,包含转录因子TBX1等重要基因。ClinGen数据库提示22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672)具有明确三倍剂量致病效应(Triplosensitivityscore:3) | arr[hg19]22q11.21(18,648,855-21,800,471)x3 | 正常 | 正常分娩 |
| 7 | 19+1 | 高龄/不良孕产史 | 女 | 胎儿染色体22q11.21区域存在1.084Mb的DNA片段缺失,涉及16个OMIM基因。ClinGen数据库提示该缺失包含22q11.2recurrentregion(central,B/C-D,chr22:20,731,986-21,465,672)(includesCRKL)区域 | arr[hg19]22q11.21(20,716,876-21,800,471)x1 | 胎儿室间隔缺损,肺动脉轻度狭窄,主动脉骑跨,胸腺偏小 | 终止妊娠 |
| 8 | 20+4 | NIPT异常/不良孕产史 | 男 | 胎儿染色体22q11.21区域存在1.084Mb的片段缺失。ClinGen数据库提示该缺失片段包含22q11.2recurrentregion(central,B/C-D)(includesCRKL)(chr22:20,731,986-21,465,672),鉴于临床表型多变、不完全外显、缺乏病例-对照数据,目前单倍剂量致病效应评分为2(Haploinsufficiencyscore:2) | arr[hg19]22q11.21(20,716,876-21,800,471)x1 | 胎儿正常 | 正常分娩 |
| 9 | 19+2 | NIPT异常 | 男 | 胎儿染色体22q11.21区域存在2.881Mb的片段缺失。ClinGen数据库提示该片段基本覆盖22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672)(Haploinsufficiencyscore:3),且>90%的缺失为新发变异 | arr[hg19]22q11.21(18,919,477-21,800,471)x1 | 右位主动脉弓,左锁骨下动脉迷走,心轴左偏73度,局限性心包积液,动脉导管走行迂曲 | 终止妊娠 |
| 10 | 26+6 | 超声异常 | 男 | 胎儿染色体22q11.21存在2.888Mb的片段缺失,含有42个OMIM基因,包含22q11.2recurrent(DGS/VCFS)region(proximal,A-D)(includesTBX1)(chr22:18,912,231-21,465,672)关键区域,Clingen提示单倍剂量致病明确 | arr[hg19]22q11.21chr22:18,912,231-21,465,672) | 胎儿室间隔缺,主动脉发育不良,主动脉弓离断(B型) | 终止妊娠 |
| 11 | 26+4 | 遗传 | 女 | 胎儿染色体22q11.21区域存在1.07Mb的DNA片段缺失,涉及14个OMIM基因。ClinGen数据库提示该缺失包含22q11.2recurrentregion(central,B/C-D,chr22:20,731,986-21,465,672)(includesCRKL)区域 | arr[hg19]22q11.21(20,730,143-21,800,471)x1 | 胎儿左肾多囊肾、胎儿右肾盆腔肾、右下肢偏短 | 终止妊娠 |
| 12 | 22+6 | 超声异常 | 男 | 胎儿染色体22q11.21存在1.664Mb的片段缺失,涉及29个OMIM基因。Clingen数据库提示该片段缺失包含22q11.2recurrent(DGS/VCFS)region(proximal,A-B)(includesTBX1)(chr22:18,912,231-20,287,208),具有明确单倍剂量致病效应(Haploinsufficiencyscore:3)。 | arr[hg19]22q11.21(18,648,855-20,312,661)x1 | 室间隔缺损型肺动脉闭锁、右位主动脉弓、心轴左偏 | 终止妊娠 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
四、妊娠结局
12 例胎儿中3 例超声检查正常胎儿顺产,随访至今未见明显异常,9 例胎儿终止妊娠并验证超声检查结果(表1)。
讨 论
22q11.2DS 是由第三、第四咽囊和第四主动脉弓发育缺陷引起的,导致多系统表现,包括:圆锥动脉干型CHD、腭咽异常、颅面畸形、骨骼异常、胃肠道和肾脏异常;甲状旁腺功能减退、T 细胞免疫缺陷性自身免疫疾病,血小板减少症、认知缺陷和精神疾病等[12],因此至今有着各种不同的名称,如Digeorge 综合征、腭-心-面综合征、Catch-22综合征等。
22q11.2 微缺失是指22 号染色体长臂近着丝粒端微片段22q11.2.21 ~q11.23 缺失。其缺失区间相对固定,缺失大小约1.5 ~3.0 Mb,87%缺失3 Mb,8%缺失1.5 Mb,2%缺失2 Mb,低于染色体核型分析分辨率的下限(5 Mb),微缺失由此得名[13]。
22q11.2 区域存在低拷贝重复序列(low copy repeat,LCR;从LCR 22-A 到LCR 22-H),在细胞分裂过程中,这些LCR 之间的非等位基因不等同源交叉重组导致微缺失和重复[14-15]。位于LCR A-B 之间的TBX1 基因控制咽弓和心室流出道的细胞增殖,影响器官的形成,并在该综合征的临床表现中发挥主要作用,甚至TBX1 基因的内致病性变体也会导致CHD,然而该区域中的一些其他基因(如CRKL、LZTR、MAPK1、VEGF 等)也被描述为在表型中具有修饰作用[16]。动物实验表明,TBX1 过表达与CHD 呈正相关,表明TBX1 可能与胎儿CHD 发生有关[17]。其他基因,如HIRA、UFD1L(位于LCR A-B 之间)和CRKL(位于LCR C-D 之间)也是被确定为与心脏畸形有关的候选基因,但它们的作用不太确定[18]。在本组病例中7 例CHD 胎儿均发生在LCR 22A-D,片段缺失均>2.5 Mb,均包含TBX1 基因。在<2.5 Mb 的片段缺失病例中表现为LCR 22C-D,包含CRKL、MAPK1 基因,1 例表现为轻症TOF、1 例表现为多囊肾和股骨偏短,其余3 例未发现异常。
心脏畸形是22q11.2DS 胎儿中最常见畸形表现,在一系列妊娠期研究报告中心脏异常占62%~95%,圆锥动脉干异常是最常见的心脏缺陷类型,发现率为62%~78%,TOF 检出率为23%~45%,其次是主动脉弓离断、永存动脉干和VSD[16,19-20]。在本组病例中心脏畸形占66.7%,圆锥动脉干畸形占58.3%,最为常见的TOF 及肺动脉狭窄或闭锁,其次是右心室双出口。在超声影像学征象上,本组病例主要表现VSD、心轴左偏、肺动脉狭窄或闭锁、主动脉骑跨,右位主动脉弓、左锁骨下动脉迷走及主动脉离断,VSD 虽然占比为41.7%,但是其作为复杂CHD 的一部分出现,本组病例VSD 未见单独出现病例,因此单纯的VSD 不能作为22q11.2DS 胎儿的有效检测指标。肺动脉狭窄或闭锁、主动脉骑跨、右位主动脉弓是圆锥动脉干畸形的组成部分,也是22q11.2DS的常见心脏异常表现。心轴异常是心脏常规检查中最先发现的异常。Vigneswaran 等[21]报道心轴增大是22q11.2DS 相关异常的独立预测因子,在本组病例中也得到证实,在复杂CHD 中心轴左偏出现3 例,占所有病例的25.0%。右位主动脉弓无论是否合并其他心脏异常均增加22q11.2DS 的风险,同时右位主动脉弓及其各种分支异常、动脉导管缺如、粗大的体肺动脉侧支等亦会增加CHD 胎儿22q11.2DS 的检出率[22-23]。在本组病例中2 例右位主动脉弓中1 例出现了左锁骨下动脉迷走。右位主动脉弓及其各种分支异常、动脉导管缺如、粗大的体肺动脉侧支等亦会增加CHD 胎儿22q11.2DS 的检出率。
22q11.2DS 中的调节胸腺发育的关键转录因子被认为是TBX1,胸腺发育不良是22q11.2DS 的一个重要表征。尽管通常不会在常规产前超声检查中进行胎儿胸腺评估,但研究表明,95%的胎儿都可以识别评估胸腺[24]。胎儿胸腺发育缺如或发育不良是22q11.2DS 超声检查的一个新发现,胎儿胸腺可以从孕15 周开始测量。Chaoui 等[25]注意到在22q11.2DS 病例中胎儿胸腺发育不全,并可以通过在心脏的三血管气管切面进行胸腺与胸腔的面积比来成功评估胎儿胸腺发育情况,胸腺发育缺如或发育不良作为22q11.2DS 的超声标志物的阳性预测值为81.8%,敏感度为90%,特异度为98.5%。同时圆锥动脉干异常与胸腺发育不良/发育不全之间的关联已被充分证明[19],在本研究中也得到了印证。3 例胎儿胸腺发育不良,占25.0%,2 例合并复杂CHD,1 例为单纯发病,3 例胎儿胸腺与胸腔的面积比明显小于实际孕周。
尽管骨骼系统异常在22q11.2DS 中表现不像胎儿CHD 和胸腺异常那样常见且典型,但22q11.2DS胎儿的产前超声上观察到的骨骼异常高达18%~25%,最常报道的畸形为胎儿足内翻,占4.7%~20.0%,观察到的骨骼异常包括脊椎异常、隆突、四肢短、多指畸形和并指畸形[16,19]。在本组病例中仅有1 例胎儿为多囊肾合并左肾多囊肾、右肾盆腔肾合并右下肢长骨偏短。下肢长骨偏短与22q11.2DS 的畸形谱目前无相关报道,根据现有文献报道胎儿足内翻可以作为22q11.2DS 检测指标[26]。
据报道,22q11.2DS 胎儿中有8%~17%发生泌尿生殖系统畸形,包括肾盂扩张和肾积水、单侧或罕见双侧肾发育不全、单侧多囊肾发育不良和输尿管扩张[17,19-25,27]。本组病例中仅有1 例(8.3%)表现为胎儿左肾多囊肾、胎儿右肾盆腔肾改变。
22q11.2DS 畸形谱还伴有其他畸形,如颜面部畸形、唇腭裂、羊水过多和宫内发育迟缓等。唇腭裂胎儿由于羊水吞咽困难常伴羊水过多,文献报道16%的胎儿(81/512)出现羊水过多,在孕晚期单独发现羊水过多的22q11.2DS 胎儿占0~8%[28]。
产前超声畸形检查是孕期进行侵入性检查的有效指征,在本研究中单纯产前超声检查异常而建议进一步CMA 检测的病例占58.3%,超声和无创检查同时检出异常而建议进一步CMA 检测的病例占8.3%,而无创检查异常建议进一步CMA 检测的病例占16.7%。产前超声筛查对22q11.2DS 的畸形谱检测具有独特优势,尤其在CHD 中圆锥动脉干畸形的检查。但本研究病例较少,病例畸形特点集中于心脏畸形和胸腺异常,心外畸形较少,唇腭裂畸形、羊水过少均未发现,有待收集更多病例进行更深入的分析。
综上所述,CHD 尤其圆锥动脉干畸形及心轴异常、胸腺发育不良、骨骼、泌尿系畸形及颜面部畸形都是22q11.2DS 的广泛畸形谱的表现,也是产前超声检查的长处所在,当产前超声筛查发现相关畸形,尤其是圆锥动脉干畸形、胸腺畸形,或此类合并心内心外多发畸形时,应积极建议进行CMA检测,为孕妇提供积极准确的产前咨询。